Overview
Generally speaking, we work on biological and biomedical problems in a data-driven manner using computational and statistical techniques to discover new knowledge. Some areas of interest to us are exemplified below. However, this is not an exhaustive list and we are interested in and work on other aspects within the field of biology. Feel free to browse through our publications and tools to learn more about current and past research interests.
Proximal Gene Regulation
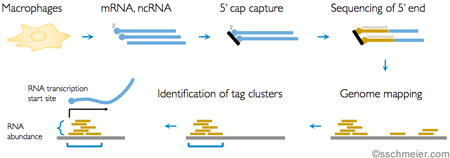 We make use of transcriptomics data stemming from technologies like RNA-seq or CAGE to understand the how genes are expressed and regulated on the transcriptional level.
For example, through CAGE (see Figure) we study 5' transcription starts sites (promoters) of RNAs and explore their transcriptional output (expression levels) in different conditions (1).
Also, we investigate if particular genes get transcribed from different promoters in particular biological conditions (alternative promoter usage), which might be related to distinct regulatory features at promoters and/or might impact splice variants of genes.
For example, we are investigating the transcriptional responses of mouse macrophages to stimulation and infection (2, 3, 4, 5).
We are also studying the use of transcriptomics profiles for cancer (sub-)type classification, as well as investigating the relationships between host transcriptomes and bacterial communities (6).
We make use of transcriptomics data stemming from technologies like RNA-seq or CAGE to understand the how genes are expressed and regulated on the transcriptional level.
For example, through CAGE (see Figure) we study 5' transcription starts sites (promoters) of RNAs and explore their transcriptional output (expression levels) in different conditions (1).
Also, we investigate if particular genes get transcribed from different promoters in particular biological conditions (alternative promoter usage), which might be related to distinct regulatory features at promoters and/or might impact splice variants of genes.
For example, we are investigating the transcriptional responses of mouse macrophages to stimulation and infection (2, 3, 4, 5).
We are also studying the use of transcriptomics profiles for cancer (sub-)type classification, as well as investigating the relationships between host transcriptomes and bacterial communities (6).
Distal Gene Regulation
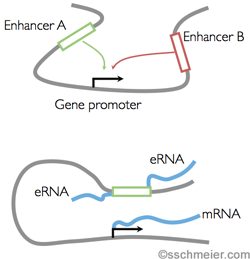 Distal cis-regulatory control can be facilitated through enhancers (see Figure).
Chromatin forms loops to bring enhancer regions physically close to a gene's promoter region.
Enhancers are known to be tissue-specific and different enhancers may drive regulation in different biological conditions.
Enhancer loci show expression of short enhancer RNAs (eRNA) who's expression correlates with the gene expression coming from the promoter.
For example, we are studying the influence of enhancers on the regulation of genes during mouse macrophage activation (7) as well as tuberculosis infection.
Enhancer chromosomal regions are characterised by particular combinations of histone modifications, which we study through the analyses of ChIP-seq datasets.
As enhancer-promoter chromosomal regions interact with each other, we also investigate data stemming from chromatin conformation capture approaches, e.g. Hi-C, 4C, etc.
Distal cis-regulatory control can be facilitated through enhancers (see Figure).
Chromatin forms loops to bring enhancer regions physically close to a gene's promoter region.
Enhancers are known to be tissue-specific and different enhancers may drive regulation in different biological conditions.
Enhancer loci show expression of short enhancer RNAs (eRNA) who's expression correlates with the gene expression coming from the promoter.
For example, we are studying the influence of enhancers on the regulation of genes during mouse macrophage activation (7) as well as tuberculosis infection.
Enhancer chromosomal regions are characterised by particular combinations of histone modifications, which we study through the analyses of ChIP-seq datasets.
As enhancer-promoter chromosomal regions interact with each other, we also investigate data stemming from chromatin conformation capture approaches, e.g. Hi-C, 4C, etc.
Non-coding RNAs
 The majority of transcribed RNAs in the mammalian genome is not coding for proteins.
These non-coding RNAs (ncRNAs) can have a variety of functions and are importnat in many key biological processes.
We are interested in understanding the functionality of ncRNAs and how they interact with one another, other mRNAs or proteins.
For example, we use network and graph theoretical as well as machine learning approaches to deduce the importance of entities within regulatory networks of biological conditions to identify key ncRNAs and proteins that e.g. exert the most influence on the network or are deregulated (8, 9, 10, 11, 12).
We study the function of long non-coding RNAs (lncRNAs) (13, 14).
LncRNAs have been implicated in many diseases.
Through collaborations we study the influence of lncRNAs on human diseases like heart disease or cancer.
The majority of transcribed RNAs in the mammalian genome is not coding for proteins.
These non-coding RNAs (ncRNAs) can have a variety of functions and are importnat in many key biological processes.
We are interested in understanding the functionality of ncRNAs and how they interact with one another, other mRNAs or proteins.
For example, we use network and graph theoretical as well as machine learning approaches to deduce the importance of entities within regulatory networks of biological conditions to identify key ncRNAs and proteins that e.g. exert the most influence on the network or are deregulated (8, 9, 10, 11, 12).
We study the function of long non-coding RNAs (lncRNAs) (13, 14).
LncRNAs have been implicated in many diseases.
Through collaborations we study the influence of lncRNAs on human diseases like heart disease or cancer.
Variations / SNPs / eQTLs
We study how DNA variations (e.g. SNPs) influence the regulation of gene expression.
For example, recently many SNPs have been identified that are located within non-coding areas of the genome but have been shown to influence the expression of genes (e.g. eQTL SNPs).
We are interested in understanding how those SNPs facilitate the observed change in expression of the associated gene(s), e.g. are those SNPs located in regions that are being bound by TFs, e.g. promoters (11) or enhancers?
Data Mining and Visualisation
Another area of research involves the identification, extraction, storage, integration, and representation of biological/biomedical data.
We are interested in data mining and machine learning techniques to harvest, mine and integrate biological data from various heterogenous sources.
We study and use database and web technologies to create knowledge-bases and web applications that help researchers find useful information and/or help analyse their data.
Examples of projects in this domain include a customised knowledge-base to study the influence of non-coding regulatory RNA on immunological processes (15), a database that helps in identifying regulatory variations in promoter regions of microRNAs (11), as well as a database that facilitates the identification of transcription co-factors and TF interactions (16, 17).
References
1. Forrest ARR, Kawaji H, Rehli M, Baillie KJ, de Hoon MJL, Haberle V, Lassmann T, Kulakovskiy IV, Lizio M, Itoh M, Andersson R, Mungall CJ, Meehan TF, Schmeier S, Bertin N, Jorgensen M, Dimont E, Arner E, Schmidl C, Schaefer U, Medvedeva YA, Plessy C, Vitezic M, Severin J, Semple C, Ishizu Y, Young RS, Francescatto M, Alam I, Albanese D, … Hayashizaki Y. A promoter-level mammalian expression atlas. Nature 2014 507 462–470. (doi:10.1038/nature13182)
2. Roy S, Guler R, Parihar SP, Schmeier S, Kaczkowski B, Nishimura H, Shin JW, Negishi Y, Ozturk M, Hurdayal R, Kubosaki A, Kimura Y, de Hoon MJL, Hayashizaki Y, Brombacher F, & Suzuki H. Batf2/Irf1 Induces Inflammatory Responses in Classically Activated Macrophages, Lipopolysaccharides, and Mycobacterial Infection. J. Immunol. 2015 194 6035–6044. (doi:10.4049/jimmunol.1402521)
3. Roy S, Schmeier S, Arner E, Alam T, Parihar SP, Ozturk M, Tamgue O, Kawaji H, de Hoon MJL, Itoh M, Lassmann T, Carninci P, Hayashizaki Y, Forrest ARR, Bajic VB, Guler R, Brombacher F, & Suzuki H. Redefining the transcriptional regulatory dynamics of classically and alternatively activated macrophages by deepCAGE transcriptomics. Nucleic Acids Res. 2015 43 6969–6982. (doi:10.1093/nar/gkv646)
4. Roy S, Guler R, Parihar SP, Schmeier S, Kaczkowski B, Nishimura H, Shin JW, Negishi Y, Ozturk M, Hurdayal R, Kubosaki A, Kimura Y, de Hoon MJL, Hayashizaki Y, Brombacher F, & Suzuki H. Batf2/Irf1 induces inflammatory responses in classically activated macrophages, LPS and mycobacterial infection. Eur. J. Immunol.2016. p 7. .
5. Guler R, Parihar SP, Savvi S, Logan E, Schwegmann A, Roy S, Nieuwenhuizen NE, Ozturk M, Schmeier S, Suzuki H, & Brombacher F. IL-4Ralpha-Dependent Alternative Activation of Macrophages Is Not Decisive for Mycobacterium tuberculosis Pathology and Bacterial Burden in Mice. PLoS One 2015 10 e0121070. (doi:10.1371/journal.pone.0121070)
6. Purcell RV, Visnovska M, Biggs PJ, Scmeier S, & Frizelle FA. Distinct gut microbiome patterns associate with consensus molecular subtypes of colorectal cancer. Sci Rep 2017 7 11590. (doi:10.1038/s41598-017- 11237-6)
7. Denisenko E, Guler R, Mhlanga M, Suzuki H, Brombacher F, & Schmeier S. Genome-wide profiling of transcribed enhancers during macrophage activation. . bioRxiv preprint 2017 . (doi:10.1101/163519)
8. Ravasi T, Suzuki H, Cannistraci CV, Katayama S, Bajic VB, Tan K, Akalin A, Schmeier S, Kanamori-Katayama M, Bertin N, Carninci P, Daub CO, Forrest ARR, Gough J, Grimmond S, Han J-H, Hashimoto T, Hide W, Hofmann O, Kamburov A, Kaur M, Kawaji H, Kubosaki A, Lassmann T, van Nimwegen E, MacPherson CR, Ogawa C, Radovanovic A, Schwartz A, Teasdale RD, … Hayashizaki Y. An atlas of combinatorial transcriptional regulation in mouse and man. Cell 2010 140 744–752. (doi:10.1016/j.cell.2010.01.044)
9. Suzuki H, Forrest ARR, van Nimwegen E, Daub CO, Balwierz PJ, Irvine KM, Lassmann T, Ravasi T, Hasegawa Y, de Hoon MJL, Katayama S, Schroder K, Carninci P, Tomaru Y, Kanamori-Katayama M, Kubosaki A, Akalin A, Ando Y, Arner E, Asada M, Asahara H, Bailey T, Bajic VB, Bauer D, Beckhouse AG, Bertin N, Björkegren J, Brombacher F, Bulger E, Chalk AM, … Hayashizaki Y. The transcriptional network that controls growth arrest and differentiation in a human myeloid leukemia cell line. Nat. Genet. 2009 41 553–562. (doi:10.1038/ng.375)
10. Schmeier S, MacPherson CR, Essack M, Kaur M, Schaefer U, Suzuki H, Hayashizaki Y, & Bajic VB. Deciphering the transcriptional circuitry of microRNA genes expressed during human monocytic differentiation. BMC Genomics 2009 10 595. (doi:10.1186/1471-2164-10-595)
11. Schmeier S, Schaefer U, MacPherson CR, & Bajic VB. dPORE-miRNA: polymorphic regulation of microRNA genes. PLoS One 2011 6 e16657. (doi:10.1371/journal.pone.0016657)
12. Schmeier S, Schaefer U, Essack M, & Bajic VB. Network analysis of microRNAs and their regulation in human ovarian cancer. BMC Syst. Biol. 2011 5 183. (doi:10.1186/1752-0509-5-183)
13. Hon CC, Ramilowski JR, Harshbarger J, Bertin N, Rackham OJL, Gough J, Denisenko E, Schmeier S, Poulsen TM, Severin J, Lizio M, Kawaji H, Kasukawa T, Itoh M, Burroughs MA, Noma S, Djebali S, Alam T, Medvedeva YA, Testa AC, Lipovich L, Yip CW, Abugessaisa I, Mendez M, Hasegawa A, Tan D, Lassmann T, Heutink P, Babina M, Wells C.A, … Forrest ARR. An atlas of human long non-coding RNAs with accurate 5’ ends. Nature 2017 543 199–204. (doi:10.1038/nature21374)
14. Salhi A, Essack M, Alam T, Bajic VP, Ma L, Radovanovic A, Marchand B, Schmeier S, Zhang Z, Uludag M, & B. BV. DES-ncRNA: A knowledgebase for exploring information about human micro and long noncoding RNAs based on literature-mining. RNA Biology 2017 . (doi:10.1080/15476286.2017.1312243)
15. Denisenko E, Ho D, Tamgue O, Ozturk M, Suzuki H, Brombacher F, Guler R, & Schmeier S. IRNdb: the database of immunologically relevant non-coding RNAs. Database 2016 baw138 1–9. (doi:10.1093/database/baw138)
16. Schmeier S, Alam T, Essack M, & Bajic VB. TcoF-DB v2: update of the database of human and mouse transcription co-factors and transcription factor interactions. Nucleic Acids Res. 2017 45 D145–D150. (doi:10.1093/nar/gkw1007)
17. Schaefer U, Schmeier S, & Bajic VB. TcoF-DB: dragon database for human transcription co-factors and transcription factor interacting proteins. Nucleic Acids Res. 2011 39 D106–D110. (doi:10.1093/nar/gkq945)